華大在線訊(通訊員 周漢鳴)4月4日,我校化學學院肖文精教授團隊在國際頂級期刊《自然催化》(Nature Catalysis)發表題為"催化的金屬偶極體接力實現對映選擇性大環化"(Enantioselective macrocyclization via catalytic metallic dipole relay)的研究論文。該研究創新性地提出了"金屬偶極體接力"策略,為軸手性大環化合物的高效合成開辟了新路徑。華中師范大學博士研究生曲寶樂(實驗研究)和肖萌(理論計算)為共同第一作者,陸良秋教授和張之涵教授為共同通訊作者,我校為第一完成單位。

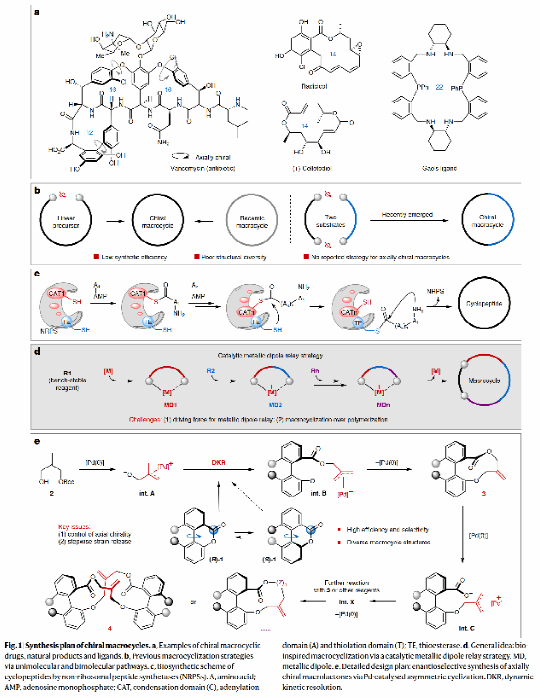
手性大環化合物作為藥物和功能材料的關鍵結構單元,其合成一直是有機化學領域的重大挑戰。以"人類抗生素最后防線"萬古霉素為例,其核心結構正是由軸手性聯芳基單元與大環內酯構成。傳統合成方法普遍面臨環化效率低、手性控制難等瓶頸問題,嚴重制約了這類化合物的開發應用。
針對這些挑戰,研究團隊從非核糖體環肽的生物合成機制獲得靈感,獨辟蹊徑,首次提出"π-烯丙基鈀偶極體接力"的新思路,通過動態動力學拆分與不對稱偶極環化的協同作用,成功實現了兩類14元、一類15元和一類20元軸手性大環內酯的高效構筑。具體而言,研究團隊首先優化了關鍵中間體手性10元環內酯的合成條件,發現帶氫鍵給體的手性雙膦配體能夠在溫和條件下獲得83%的分離收率和90%的對映選擇性(ee)。在此基礎上,研究了該催化體系在不對稱[6+4]環化反應中的普適性,并成功實現了分步的[6+4]和[10+10]串聯環化反應,高選擇性地構建了軸手性的20元大環內酯化合物(高達>99% ee)。在動力學實驗與DFT計算等機理研究的基礎上,研究團隊提出了截斷(10+10)偶極環化、發展(10+x)偶極環化的設想,成功實現了軸手性10元環內酯與氮雜二烯、α-重氮酮以及乙烯基環丙烷的一系列不對稱(10+4)(10+2+2)和(10+5)偶極環化反應,合成了結構多樣的14元和15元軸手性大環內酯化合物。甚至從最初的聯芳基6元環內酯出發,實現一步或一鍋的不對稱(6+4)/(10+x)串聯環化反應,顯著提升了上述4類軸手性大環內酯的合成效率。研究團隊通過理論計算,成功揭示了聯芳基二面角及內酯鍵與芳基二面角的變化規律,證明了聯芳基扭轉張力的逐步釋放是成功的關鍵。
該研究發展的"金屬偶極體中繼策略"不僅顯著提升了手性大環內酯的合成效率,還兼具產物結構多樣、官能團兼容性好、立體選擇性高的優勢,為手性大環化合物的不對稱催化合成提供了全新思路。
陸良秋教授長期致力于手性雜環不對稱合成研究。針對雜環合成領域長期存在的環系大小與立體選擇性控制難題,系統性地開展了金屬催化的不對稱偶極環化反應研究,為開發抗癌活性分子和腫瘤細胞成像功能分子提供了重要平臺和工具,近期在《美國化學會志》(J. Am. Chem. Soc. 2025, 147, 3223; 2024, 146, 26622-26629; 2022, 144, 19932-19941)和《德國應用化學》(Angew. Chem. Int. Ed. 2024, 63, e202319728; 2024, 63, e202408426; 2023, 62, e202301592; 2023, 62, e202212444; 2022, 61, e202117215)等頂級化學專業期刊上發表多篇研究論文。相關研究工作長期得到國家自然科學基金委、湖北省科技廳、華中師范大學、武漢光化學技術研究院、河南師范大學和中國科學院蘭州化學物理研究所等機構的經費支持。
論文鏈接:https://www.nature.com/articles/s41929-025-01322-9
(審讀人:郭彥炳 段治國)
 Copyright ? 2005-2020版權所有:華中師范大學 鄂ICP備05003325號-9
Copyright ? 2005-2020版權所有:華中師范大學 鄂ICP備05003325號-9 鄂公網安備 42011102000286號
鄂公網安備 42011102000286號







